Test de Portadores de Enfermedades Genéticas Recesivas
¿Qué es la detección de portadores de mutaciones de enfermedades recesivas?
La detección de portadores es una prueba genética que identifica mutaciones en genes relacionados con enfermedades recesivas, desarrollada para parejas en planificación familiar.
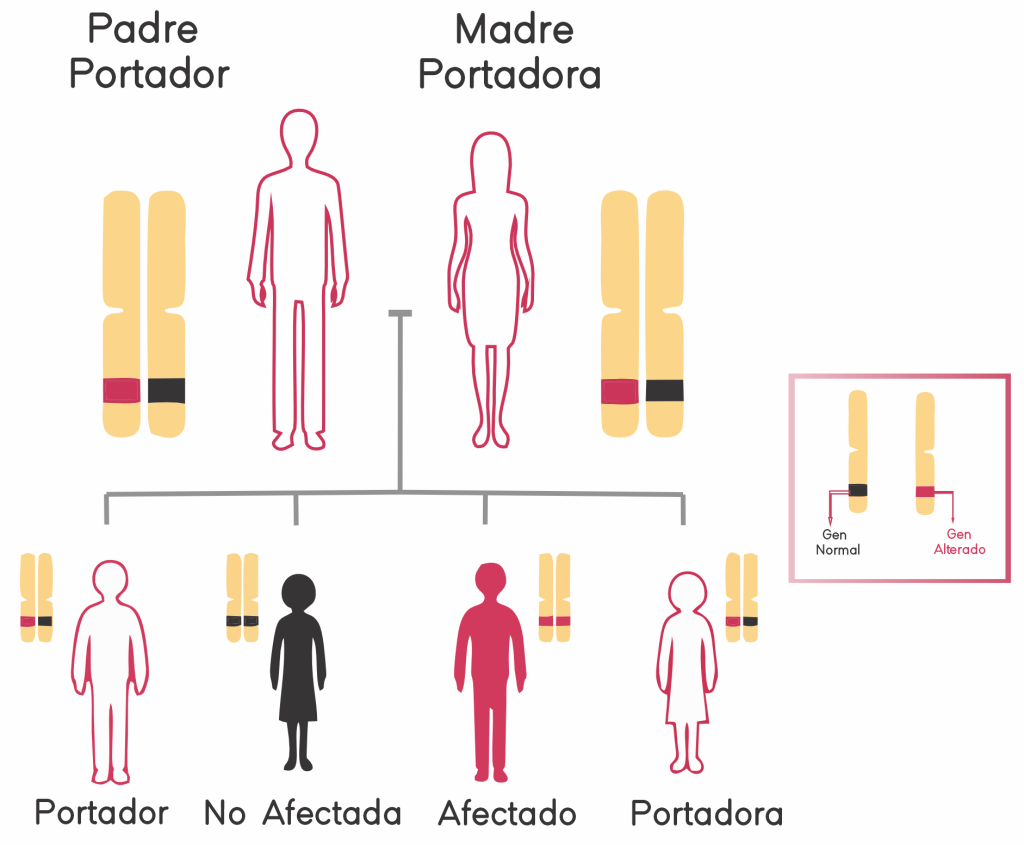
Comprenda cómo las enfermedades con un patrón de herencia recesivo se comparten en una familia.
¿Qué esperar del resultado de este examen?
Si se hacen pruebas a ambos miembros de la pareja, el resultado puede proporcionar información sobre el riesgo de tener un hijo con una condición genética recesiva.
Si el resultado indica que la pareja tiene una mutación en el mismo gen, el riesgo de tener un hijo con enfermedad genética es al menos del 25%. En este caso, la pareja puede optar por la fecundación in vitro (FIV) y el diagnóstico genético preimplantacional (DGP).
¿Cuándo y quién puede realizar el examen?
- Parejas en planificación familiar
- Personas con mayor riesgo de enfermedades recesivas
- Clínicas de reproducción que trabajan con donantes de esperma y óvulos
Esta prueba debe usarse como una herramienta de asesoramiento genético para las personas con mayor riesgo de tener una mutación por enfermedades recesivas.
Indicado principalmente para familias formadas por parejas consanguíneas o para poblaciones con mayor riesgo de enfermedades recesivas, como la población judía, principalmente de herencia Ashkenazi, pero sin limitarse a ella, pudiendo extenderse a otras poblaciones como Sefaradim y Mizrahim, por ejemplo.
Test genético antes de quedar embarazada la pareja, útil en la planificación de tener una familia, examina el riesgo de concebir un niño con una enfermedad genética.
Es importante saber que en caso se detecte que ambos miembros de la pareja familiar tengan el mismo gen afectado, la descendencia tendría un 25% de probabilidad de padecer la enfermedad genética recesiva asociada.
¿Qué es un PORTADOR?
Un portador, en relación con la genética, es una persona que posee una sola copia afectada de un gen, o sea “porta” y puede transmitir a sus hijos una variante genómica (alélica) asociada con una enfermedad (o rasgo) que se hereda en forma autosómica recesiva o en relación con el sexo, y no presenta síntomas de esa enfermedad (o características de ese rasgo). El portador ha heredado la variante alélica de un progenitor y un alelo normal del otro progenitor. Los hijos de los portadores tienen a su vez riesgo de heredar una variante alélica de sus progenitores, lo que les llevaría a tener la misma enfermedad (o rasgo).
Existe gran variedad de enfermedades de causa genética, que pueden ser transmitidas de una generación a otra, más de 10,000 enfermedades genéticas descritas en el mundo. Muchas veces una pareja es portadora de una enfermedad de este tipo sin saberlo y toman conocimiento recién al tener un hijo afectado.
Transmisión del PORTADOR
Para entender el significado de la palabra «portador» debemos recordar que cada individuo posee dos copias de cada gen. Un portador es un individuo que tiene un cambio en una de esas dos copias. Cuando dos individuos que son portadores se encuentran y tienen descendencia, si esa descendencia hereda el cambio o la copia defectuosa del gen de cada progenitor, ese individuo, los hijos, sufrirá un desorden recesivo y tendrá las características clínicas de una enfermedad recesiva. En el pasado se consideró que los portadores, o individuos que llevaban una copia incorrecta de un gen, no mostraban signos clínicos. No obstante, cada vez hay más evidencia de que en algunas enfermedades recesivas los portadores sí manifiestan de hecho algunos síntomas clínicos, y es algo que continúa descubriéndose.
Según la Organización Mundial de la Salud (OMS), entre el 4% y 7% de la población mundial sufren de enfermedades genéticas. En Perú, cerca de 1.4 millones de personas estarían padeciéndolas.
En ese marco, es importante precisar que, estas enfermedades son graves y en conjunto son responsables del 35% de las muertes durante el primer año de vida de los pacientes que las portan.
Identificando al PORTADOR
- Persona que posee una sola copia afectada de un gen.
- No presenta síntomas ni desarrolla enfermedad.
- Su descendencia sí puede desarrollar la enfermedad genética.
- Puede no registrar antecedentes familiares.
La importancia del examen previo
Es importante que se pueda estudiar a la pareja previo a la concepción para determinar si alguno de sus miembros es portador de una enfermedad recesiva y se puedan plantear alternativas para poder tener un bebé sano en casa.
Las enfermedades genéticas, como conjunto, son responsables del 35% de muertes en el primer año de vida.
Se estima que cualquier persona es portadora de entre 100 a 300 variantes que podrían ser dañinas y que todos podemos ser portadores en promedio de 20 enfermedades recesivas.
Planificación de la familia
Este test nos permite determinar que mutaciones genéticas tiene cada persona y la compatibilidad con su pareja. La prueba identifica si los padres son portadores de una o más mutaciones genéticas recesivas. Analiza más de 600 enfermedades y 14.000 mutaciones. Se trata de una prueba genética importante en la planificación de una familia, ya que ayuda a determinar el riesgo de concebir un niño con una enfermedad genética.
Los trastornos que evaluamos son mayormente autosómicos recesivos. La mayoría de las personas no saben que son portadoras hasta que se han sometido a la detección de portadores porque generalmente no tienen síntomas.
Si ambos padres son portadores del mismo trastorno, existe una probabilidad de 1 en 4 de que el bebé herede el trastorno.
¿Qué es una enfermedad genética recesiva?
La información que rige nuestras características y gobierna nuestro funcionamiento está contenida en unidades de ADN o genes. Todo ser humano posee 2 copias de cada gen, una heredada de su padre y la otra de su madre. Los genes no son iguales en todas las personas, pues existen variaciones responsables de la variabilidad genética de nuestra especie; sin embargo, existen variantes que alteran el normal funcionamiento de los genes y por ende se les considera mutaciones patogénicas. Cuando una enfermedad se da por alteración o mutación de un solo tipo de gen, se denomina enfermedad monogénica, las cuales se dividen en dos tipos: dominantes y recesivas.
Las enfermedades genéticas recesivas aparecen cuando las dos copias de un gen tienen una mutación patogénica. Para que suceda esto, ambos progenitores han tenido que ser portadores de una mutación, casi siempre sin saberlo.
Las enfermedades genéticas recesivas no tienen cura, por ello es vital poder diagnosticarlas de manera precoz para poder darle calidad de vida al paciente con un tratamiento oportuno. Dentro de este grupo de enfermedades tenemos a la fibrosis quística, fenilcetonuria, hemofilia, mucopolisacaridosis, talasemias, entre otras.
Lista de enfermedades analizadas
| Enfermedades | Genes |
| Enfermedad de Wilson | ATP7B |
| Deficiencia primaria de carnitina | SLC22A5 |
| Fenilcetonuria | PAH |
| Hiperfenilalaninemia, deficiencia de BH4, A | PTS |
| Acidemia metilmalónica relacionada con el gen MUT | MUT |
| Acidemia metilmalónica relacionada con el gen MMAA | MMAA |
| Acidemia metilmalónica relacionada con el gen MMAB | MMAB |
| Aciduria metilmalónica y homocistinuria tipo cblC | MMACHC |
| Aciduria metilmalónica y homocistinuria tipo cblD | MMADHC |
| Homocistinuria-anemia megaloblástica tipo cblE | MTRR |
| Homocistinuria-Anemia megaloblástica tipo cblG | MTR |
| Acidemia metilmalónica relacionada con MCEE | MCEE |
| Homocistinuria debida a deficiencia de cistationina beta-sintasa | CBS |
| Acidemia glutárica I | GCDH |
| Acidemia glutárica IIA | ETFA |
| Acidemia glutárica IIB | ETFB |
| Acidemia glutárica IIC | ETFDH |
| Deficiencia de acil-CoA deshidrogenasa de cadena media | ACADM |
| Deficiencia de acil-CoA deshidrogenasa de cadena corta | ACADS |
| Deficiencia de acil-CoA deshidrogenasa de cadena muy larga | ACADVL |
| Deficiencia de 3-metilcrotonil-CoA carboxilasa 1 | MCCC1 |
| Deficiencia de 3-metilcrotonil-CoA carboxilasa 2 | MCCC2 |
| Citrulinemia | ASS1 |
| Acidemia isovalérica | IVD |
| Propionicacidemia | PCCA, PCCB |
| Enfermedad por almacenamiento de glucógeno tipo Ia | G6PC |
| Enfermedad por almacenamiento de glucógeno tipo Ib | SLC37A4 |
| Enfermedad por almacenamiento de glucógeno tipo Ic | SLC37A4 |
| Enfermedad por almacenamiento de glucógeno tipo II | GAA |
| Enfermedad de almacenamiento de glucógeno tipo IV | GBE1 |
| Enfermedad de Niemann-Pick tipo A | SMPD1 |
| Enfermedad de Niemann-Pick tipo B | SMPD1 |
| Enfermedad de Niemann-Pick tipo C1 | NPC1 |
| Enfermedad de Niemann-Pick tipo C2 | NPC2 |
| Enfermedad de la orina con olor a jarabe de arce tipo 1A | BCKDHA |
| Enfermedad de la orina con olor a jarabe de arce tipo 1B | BCKDHB |
| Enfermedad de la orina con olor a jarabe de arce tipo 2 | DBT |
| Enfermedad de la orina con olor a jarabe de arce tipo 3 | DLD |
| Síndrome de Hurler | IDUA |
| Síndrome de Hurler-Scheie | IDUA |
| Mucopolisacaridosis tipo V | IDUA |
| Mucopolisacaridosis II | IDS |
| Mucopolisacaridosis tipo IIIA | SGSH |
| Mucopolisacaridosis tipo IIIB | NAGLU |
| Mucopolisacaridosis tipo IIIC | HGSNAT |
| Mucopolisacaridosis tipo IIID | GNS |
| Mucopolisacaridosis tipo IVA | GALNS |
| Mucopolisacaridosis tipo IVB | GLB1 |
| Mucopolisacaridosis tipo VI | ARSB |
| Tirosinemia tipo 1 | FAH |
| Enfermedad de Fabry | GLA |
| Deficiencia de biotinidasa | BTD |
| Deficiencia de holocarboxilasa sintetasa | HLCS |
| Síndrome de hiperornitinemia-hiperamonemia-homocitrulinuria | SLC25A15 |
| Deficiencia de carbamoilfosfato sintetasa I | CPS1 |
| Deficiencia de ornitina transcarbamilasa | OTC |
| Aciduria argininosuccínica | ASL |
| Encefalopatía por glicina | AMT, GLDC |
| Deficiencia de 3-hidroxi-3-metilglutaril-CoA sintasa 2 | HMGCS2 |
| Trastornos congénitos de la glicosilación Ia | PMM2 |
| Trastorno de la biogénesis del peroxisoma 1A (Zellweger) | PEX1 |
| Enfermedad de Krabbe | GALC |
| Hipoglucemia hiperinsulinémica familiar 2 | KCNJ11 |
| Hipoglucemia hiperinsulinémica familiar 4 | HADH |
| Hipofosfatasia infantil | ALPL |
| Hipofosfatasia, infancia | ALPL |
| Leucodistrofia metacromática por arilsulfatasa A | ARSA |
| Galactosemia | GALT |
| Alfa-manosidosis | MAN2B1 |
| Deficiencia de beta-cetotiolasa | ACAT1 |
| Deficiencia de adenosina desaminasa | ADA |
| Sitosterolemia | ABCG5, ABCG8 |
| Deficiencia del cofactor A del molibdeno | MOCS1 |
| Intolerancia hereditaria a la fructosa | ALDOB |
| Enfermedad de Tay-Sachs | HEXA |
| Síndrome de Smith-Lemli-Opitz | DHCR7 |
| Distrofia muscular de Duchenne | DMD |
| Atrofia muscular espinal | SMN1 |
| Síndrome de Joubert 2 | TMEM216 |
| Síndrome de Joubert 3 | AHI1 |
| Síndrome de Joubert 5 | CEP290 |
| Síndrome de Joubert 6 | TMEM67 |
| Síndrome de Joubert 9 | CC2D2A |
| Síndrome de Joubert 17 | C5orf42 |
| Miopatía centronuclear ligada al cromosoma X | MTM1 |
| Ceroidolipofuscinosis neuronal 1 | PPT1 |
| Enfermedades | Genes |
| Cero-lipofuscinosis neuronal 2 | TPP1 |
| Cero-lipofuscinosis neuronal 3 | CLN3 |
| Cero-lipofuscinosis neuronal 4A | CLN6 |
| Ceroidolipofuscinosis neuronal 5 | CLN5 |
| Ceroidolipofuscinosis neuronal 6 | CLN6 |
| Ceroidolipofuscinosis neuronal 7 | MFSD8 |
| Distrofia muscular de cintura tipo 2A | CAPN3 |
| Distrofia muscular de cintura tipo 2B | DYSF |
| Distrofia muscular de cintura tipo 2C | SGCG |
| Distrofia muscular de cintura tipo 2D | SGCA |
| Leucoencefalopatía megalencefálica con quistes subcorticales 1 | MLC1 |
| Enfermedad de Canavan | ASPA |
| Osteopetrosis autosómica recesiva 1 | TCIRG1 |
| Albinismo oculocutáneo tipo 1 | TYR |
| Albinismo oculocutáneo tipo 2 | OCA2 |
| Albinismo oculocutáneo tipo 3 | TYRP1 |
| Albinismo oculocutáneo tipo 4 | SLC45A2 |
| Albinismo oculocutáneo tipo 6 | SLC24A5 |
| Albinismo oculocutáneo tipo 7 | C10orf11 |
| Albinismo ocular ligado al cromosoma X | GPR143 |
| Síndrome de Hermansky-Pudlak 1 | HPS1 |
| Síndrome de Hermansky-Pudlak 3 | HPS3 |
| Ictiosis congénita autosómica recesiva 1 | TGM1 |
| Ictiosis congénita autosómica recesiva 4A | ABCA12 |
| Ictiosis congénita autosómica recesiva 4B | ABCA12 |
| Síndrome de Netherton | SPINK5 |
| Síndrome de Sjögren-Larsson | ALDH3A2 |
| Epidermólisis ampollosa de unión relacionada con el gen LAMA3 | LAMA3 |
| Epidermólisis ampollosa juntural relacionada con el gen LAMB3 | LAMB3 |
| Epidermólisis ampollosa juntural relacionada con el gen LAMC2 | LAMC2 |
| Epidermólisis ampollosa juntural tipo no Herlitz | COL17A1 |
| Epidermólisis ampollosa distrófica autosómica recesiva | COL7A1 |
| Hemofilia B | F9 |
| Alfa-talasemia | HBA1, HBA2 |
| Beta-talasemia | HBB |
| Anemia de células falciformes | HBB |
| Anemia de Fanconi, grupo de complementación A | FANCA |
| Anemia de Fanconi, grupo de complementación C | FANCC |
| Anemia de Fanconi, grupo de complementación D2 | FANCD2 |
| Anemia de Fanconi, grupo de complementación G | FANCG |
| Anemia de Fanconi, grupo de complementación I | FANCI |
| Linfohistiocitosis hemofagocítica familiar, 2 | PRF1 |
| Linfohistiocitosis hemofagocítica familiar, 3 | UNC13D |
| Linfohistiocitosis hemofagocítica familiar, 4 | STX11 |
| Linfohistiocitosis hemofagocítica familiar, 5 | STXBP2 |
| Síndrome de Omenn | RAG1, RAG2 |
| Inmunodeficiencia combinada grave, células B negativas | RAG1, RAG2 |
| Inmunodeficiencia combinada grave ligada al cromosoma X | IL2RG |
| Hipoplasia suprarrenal congénita ligada al cromosoma X | NR0B1 |
| Colestasis intrahepática familiar progresiva tipo 2 | ABCB11 |
| Colestasis intrahepática familiar progresiva 3 | ABCB4 |
| Colestasis intrahepática familiar progresiva 4 | TJP2 |
| Síndrome de Alport 2, autosómico recesivo | COL4A3, COL4A4 |
| Síndrome nefrótico tipo 1 | NPHS1 |
| Nefronoptisis 3 | NPHP3 |
| Nefronoptisis 11 | TMEM67 |
| Cistinosis nefropática | CTNS |
| Fibrosis quística | CFTR |
| Sordera autosómica recesiva 1A | GJB2 |
| Sordera autosómica recesiva 4, con vestibular agrandado Acueducto | SLC26A4 |
| Síndrome de Wolfram 1 | WFS1 |
| Síndrome de Ellis-van Creveld | EVC2 |
| Síndrome de osteoporosis-pseudoglioma | LRP5 |
| Síndrome de Inmunodeficiencia-inestabilidad centromérica-anomalías faciales 1 | DNMT3B |
| Síndrome de Meckel 2 | TMEM216 |
| Síndrome de Meckel 3 | TMEM67 |
| Síndrome de Meckel 4 | CEP290 |
| Displasia ectodérmica hipohidrótica ligada al cromosoma X | EDA |
| Síndrome COACH | TMEM67, CC2D2A |
| Miopatía nemalínica 2 | NEB |
| Aspartilglucosaminuria | AGA |
| Poliquistosis renal | PKHD1 |
| Disautonomía familiar | IKBKAP |
| deficiencia de tirosina hidroxilasa | TH |
| Ataxia-telangiectasia | ATM |
| Deficiencia de alfa-1 antitripsina | SERPINA1 |
| Paraplejía espástica 11, autosómica recesiva | SPG11 |
| Síndrome de Bloom | BLM |
| Fiebre mediterránea familiar | MEFV |
| Síndrome de Gitelman | SLC12A3 |
| deficiencia de galactoquinasa | GALK1 |
| Mucolipidosis IV | MCOLN1 |
| Deficiencia de glucosa-6-fosfato deshidrogenasa | G6PD |
| Argininemia | ARG1 |
| Deficiencia de 3-hidroxiacil-CoA deshidrogenasa de cadena larga | HADHA |
| Deficiencia de proteína trifuncional | HADHA, HADHB |
Más tipos de pruebas:

Secuenciación Completa del Exoma
Brinda información sobre algún desorden genético, analizando más de 20,000 genes asociados a enfermedades.

Panel Hematológico - Trombofilias
Test que analiza el riesgo de padecer trombofilias hereditarias.